

Abstract
Sickle cell disease (SCD) is an inherited hemoglobinopathy characterized by hemolysis and intermittent acute pain with multi-organ damage. Previously, we showed that acute pain in SCD was associated with >10-fold increases in cell-free DNA (cfDNA) when compared to steady state, that were significantly reduced during hydroxyurea therapy. Apoptosis, necrotic cell death and lysis of intact cells in the blood stream have been proposed as sources of plasma cfDNA. Here, we explored if the cfDNA increases could have a role in inflammation, a constant pathological feature of SCD.
cfDNA was extracted using QIAamp MinElute ccfDNA Kit (Qiagen), from the platelet-poor plasma processed within 30 minutes from the blood drawn in EDTA tubes, and analyzed using whole genome sequencing (WGS) and targeted quantitative PCR (qPCR). SCD patients are defined as in acute pain if there is no evident cause other than SCD, for which the patient needs hospitalization, either as in- or outpatient, and is treated with parenteral narcotics. Steady state was defined as the period from at any time 8 weeks prior to or after a crisis.
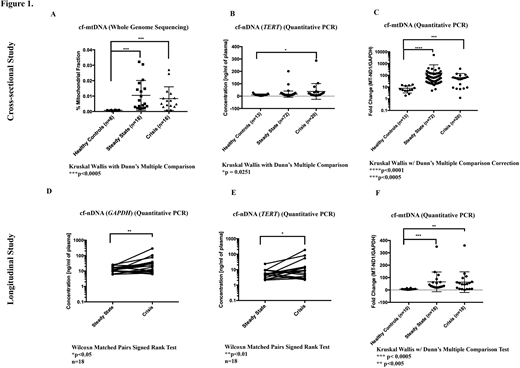
A cross-sectional study of 8 healthy controls and 34 SCD patients (18 steady-state; 16 crisis) mapped WGS reads showed significantly higher proportion of cell-free mitochondrial DNA (cf-mtDNA) compared to nuclear cfDNA (cf-nDNA) in SCD patients compared with healthy controls (Fig 1A: steady-state: 14 fold; crisis: 11 fold; p = 0.0001). We used targeted qPCR to quantify both cf-nDNA and cf-mtDNA in another cross-sectional cohort of 13 healthy controls and 92 patients (72 steady-state, 20 crisis) as well as 18 paired HbSS patients (steady-state and crisis) samples with 10 healthy controls. The nuclear reference genes used were GAPDH and TERT and mitochondrial genes were MT-ND1 and MT-ND6. While cf-nDNA (TERT) was significantly increased (> 3.5 fold, p = 0.0251; Fig 1B) in SCD patients compared with healthy controls only during crises, significantly higher levels of cf-mtDNA over cf-nDNA were observed in SCD patients compared with healthy volunteers in both steady-state and crises (Fig 1C: MT-ND1/GAPDH: steady-state >19 fold, crisis > 8 fold; MT-ND1/TERT: steady-state > 8 fold, crisis > 7 fold; MT-ND6/GAPDH: steady-state > 7 fold, crisis > 3 fold; MT-ND6/TERT: steady-state > 4 fold; crisis > 4 fold; p < 0.05).
In the paired samples, cf-nDNA (GAPDH andTERT) was significantly increased (> 3 fold; Fig 1D-E) in crisis compared to steady-state (p < 0.05). The differential increase in cf-mtDNA (cf-mtDNA:cf-nDNA ratio) levels in these patients during crises, were significantly higher compared with healthy controls (Fig 1F: MT-ND1/GAPDH: steady-state >9 fold, crisis > 8 fold; MT-ND1/TERT: steady-state > 8 fold, crisis > 9 fold; MT-ND6/GAPDH: steady-state > 8 fold, crisis > 8 fold; MT-ND6/TERT: steady-state > 8 fold; crisis > 7 fold; p < 0.005). Using confocal microscopy and mitochondrial-specific dyes (MitoTracker Green and TMRM), we show that substantial numbers of red blood cells from SCD patients retain their mitochondria in the circulation.
We next explored if the elevated cf-mtDNA in SCD could contribute to its pathophysiology, via activating neutrophils to form neutrophil extracellular traps (NETs), a recognized immunological response in inflammation. Initially, we confirmed that mtDNA can induce NETosis by treating neutrophils from healthy donors with mtDNA isolated from human platelets. mtDNA consistently induced a robust NETs response (N=8) while genomic nuclear DNA did not cause any NETosis. SCD plasma containing high levels of cf-mtDNA also caused a strong NETosis response while plasma from healthy donors did not (N=11). Cytosolic adaptor STING has a central role in sensing of cytosolic double stranded DNA. We sought to determine if the downstream STING-TBK1-IRF3 pathway is associated with the mtDNA-mediated formation of NETs. We inhibited the catalytic activity of the STING downstream effector TBK1 with BX795 prior to treating neutrophils with cf-mtDNA-containing plasma (N=5). The TBK1 inhibition consistently reduced the NETs response by at least 70% confirming that cytosolic DNA sensors are involved in promoting mtDNA-mediated formation of NETs.
Our findings suggest that cf-mtDNA induces NETosis contributing to the pathological sterile inflammation in SCD patients. Continual release of these mitochondrial DAMPs in hemolysis may serve as key link between inflammation and organ damage in SCD.
No relevant conflicts of interest to declare.

